Prepare 2 minimization in files, 3 MD in files
The third file needed by AMBER is an input parameter file (*.in). This gives AMBER information about how you will be simulating your molecule. The information includes the system pressure, temperature, non-bonded interaction distance, which atoms will be frozen during the simulation, etc. You will need to look in the AMBER manual to get the full names for these abbreviated settings in order to include them in your report.
Note that the first character of each line of each file(*.in) is a space.
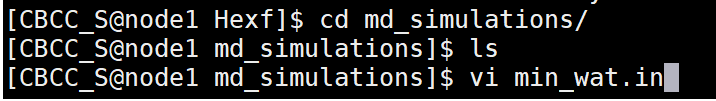
Solvent minimization (min_wat.in)
 |
|
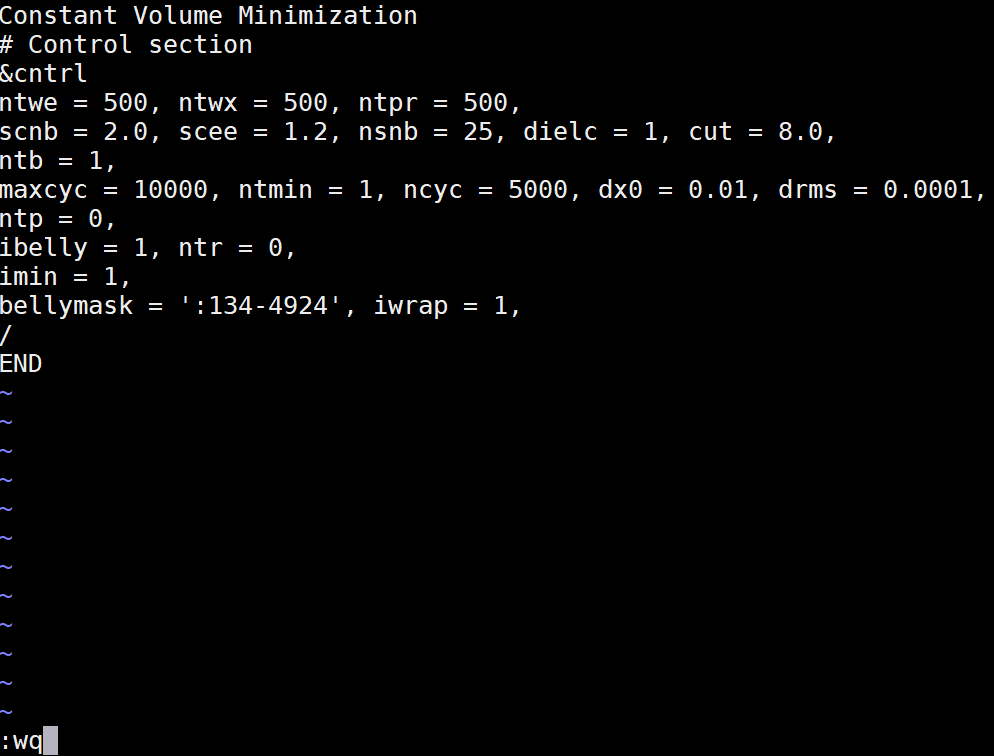 |
Copy the following text and paste it into the min_wat.in file. Edit the bellymask to contain your correct water residue range. These are the residues which are allowed to move in the minimization. Save this file to your md_simulations directory. Constant Volume Minimization |
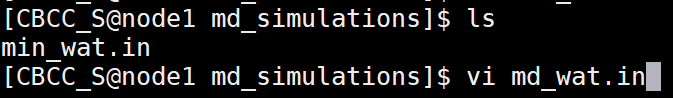
Solvent MD - simulated annealing (md_wat.in)
 |
|
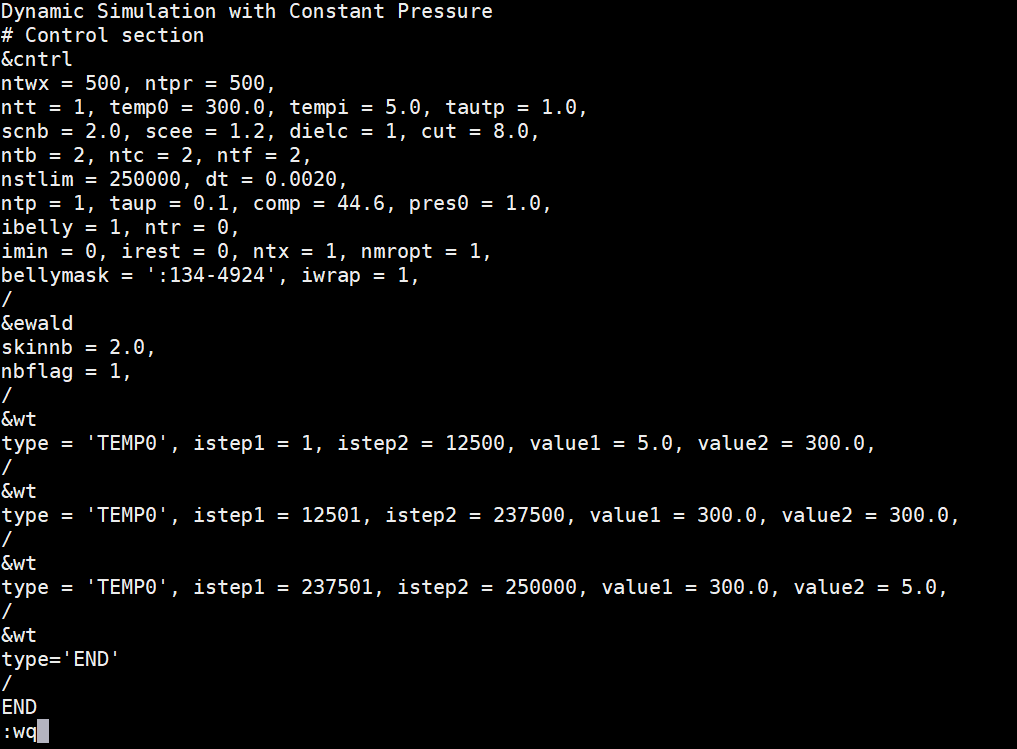 |
Copy the following text and paste it into the md_wat.in file. Edit the bellymask to contain your correct water residue range. These are the residues which are allowed to move in the MD simulation. Save this file to your md_simulations directory. Dynamic Simulation with Constant Pressure |
System minimization (min_all.in)
 |
|
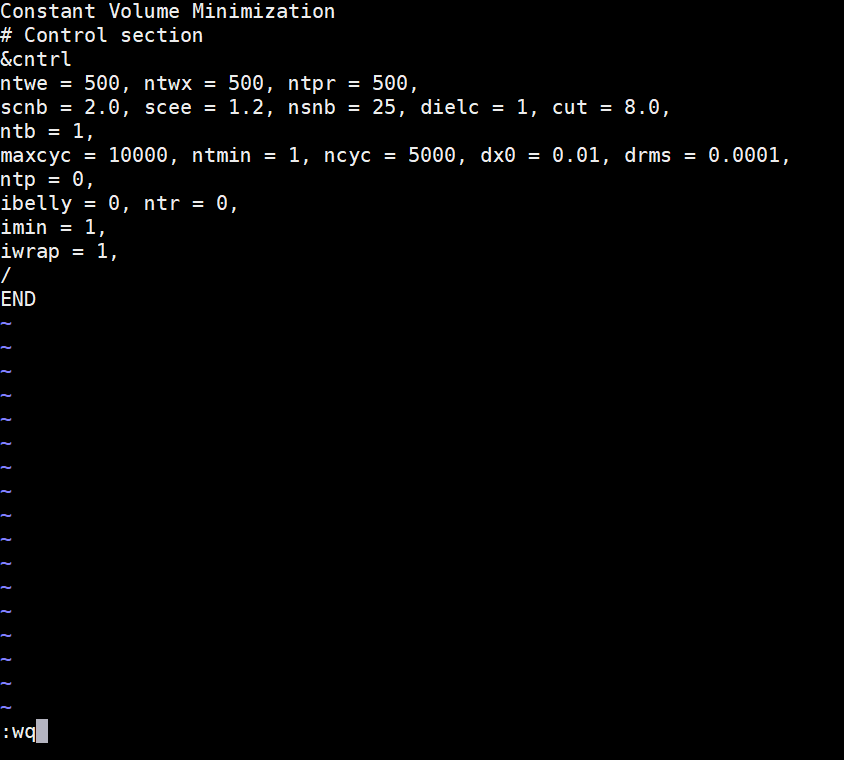 |
Copy the following text and paste it into the min_all.in file. Since all residues are allowed to move, there is no bellymask to edit. Save this file to your md_simulations directory. Constant Volume Minimization
|
System MD - production run (md_all.in)
 |
|
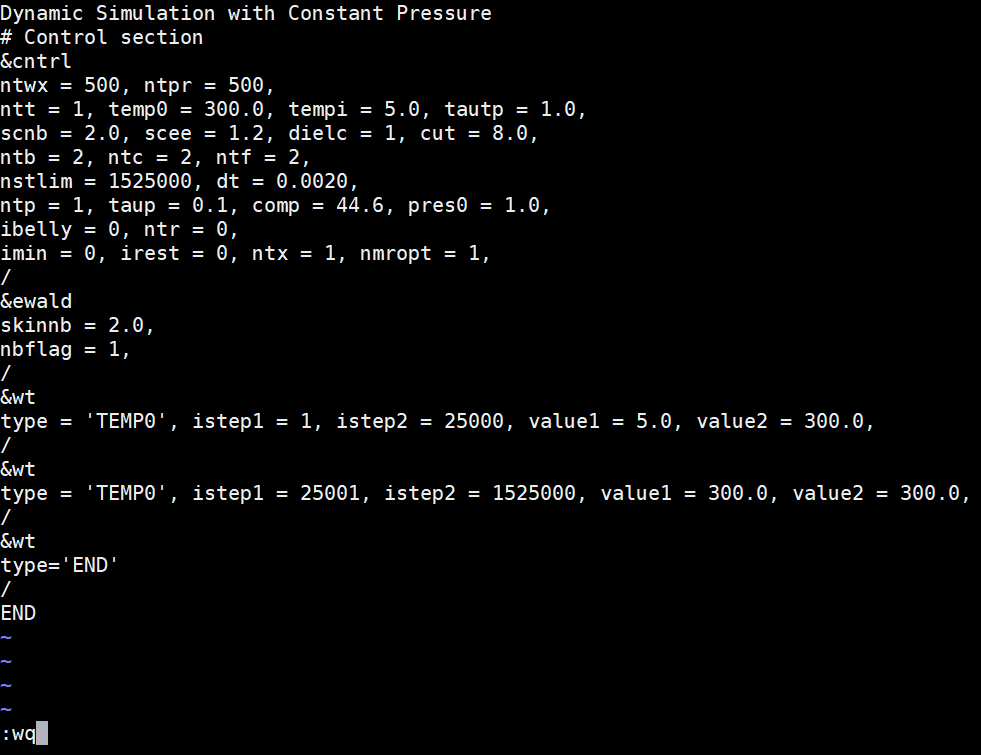 |
Copy the following text and paste it into the md_all.in file. Since all residues are allowed to move, there is no bellymask to edit. Save this file to your md_simulations directory. Dynamic Simulation with Constant Pressure
|
Sidechain/solvent MD - production run (md_back.in)
 In a terminal window, use the vi editor do open a new file called md_back.in. This will be the input parameter file for a MD simulation of the sidechains and solvent in the solvated homology model system. In this case, the sidechains and the solvent are allowed to move, but the protein backbone is restrained. The system is heated from 5K to 300K over 50ps, and held at 300K for a production run of 750ps. |
|
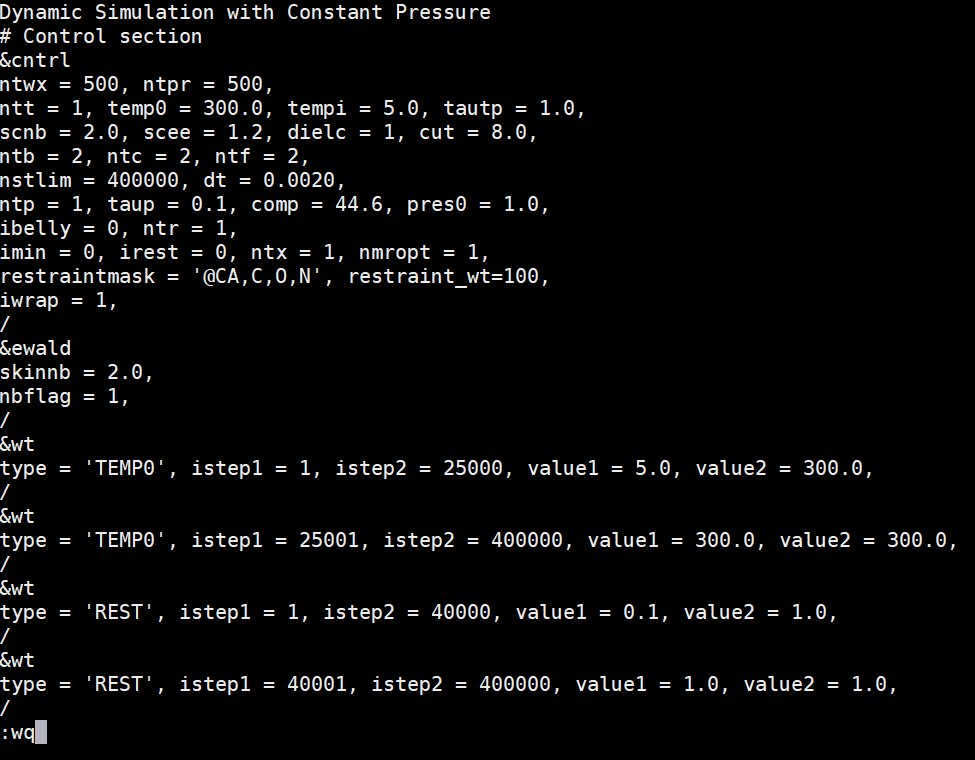 |
Copy the following text and paste it into the md_back.in file. The restraintmask is valid for all homology model simulations because it calls all backbone atoms instead of specific residues. Save this file to your md_simulations directory. Dynamic Simulation with Constant Pressure |
Prepare submission executable
At this point, all steps of the minimizations, simulated annealing, and production runs will be specified in a single executable file that will allow each command to be run consecutively. Minimizations feed into MD simulations as follows:

Comments in the executable are signified by the # sign and are not read or executed. Note that the same topology file is used in all the commands. This is done intentionally because the topology file doesn't change between simulations. The coordinate file, however, contains the position of the protein, waters and counter-ion at regular time intervals and changes for each step. Run #s 4 & 5 use the same input files from run #3 because both are doing the same thing, only #5 keeps the backbone frozen and runs for a shorter time period.
 |
|
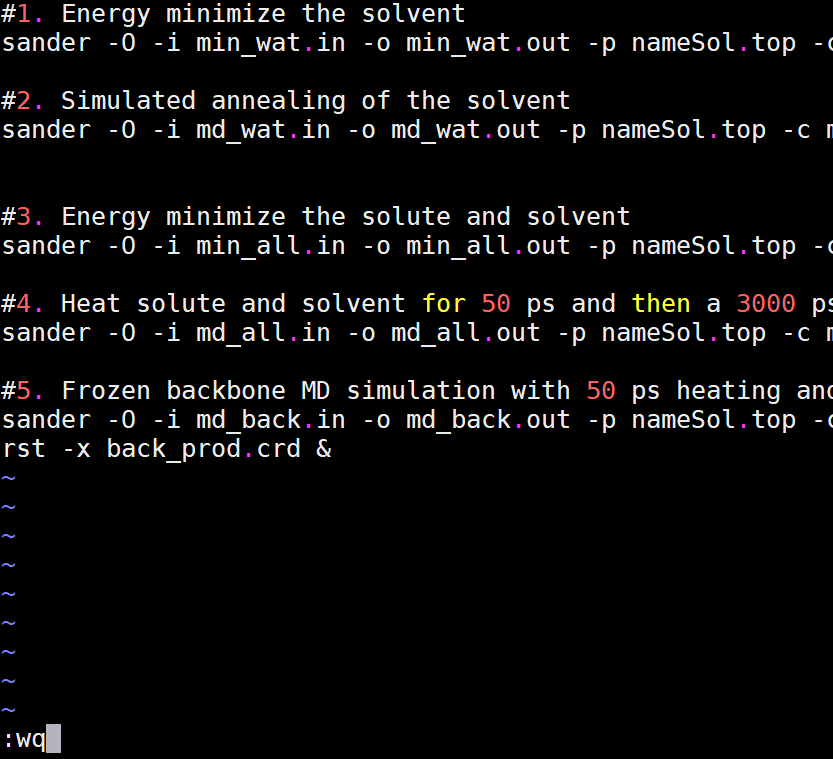 |
Copy the following text and paste it into the run.com file. Make sure all the file names match the names of your files (i.e., nameSol.top and nameSol.crd need to be changed to match the names of your topology and initial coordinate file). Save this file to your md_simulations directory. Color legend: #1. Energy minimize the solvent
|
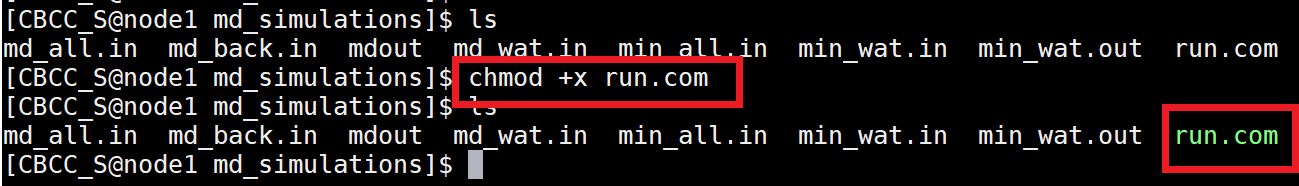 Make the run.com file executable by entering the following into the command line: $ chmod +x run.com
|
Submit simulations to run
You will now submit your simulation jobs to run on your workstation. The nohup command ensures that when you log out of your workstation today, your jobs will continue to run as long as the computer is not shut down.
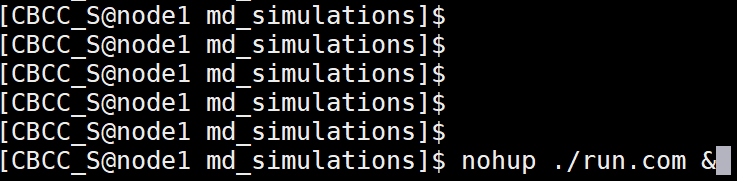 |
Enter the following at the command line to submit simulation jobs. $ nohup ./run.com &
|
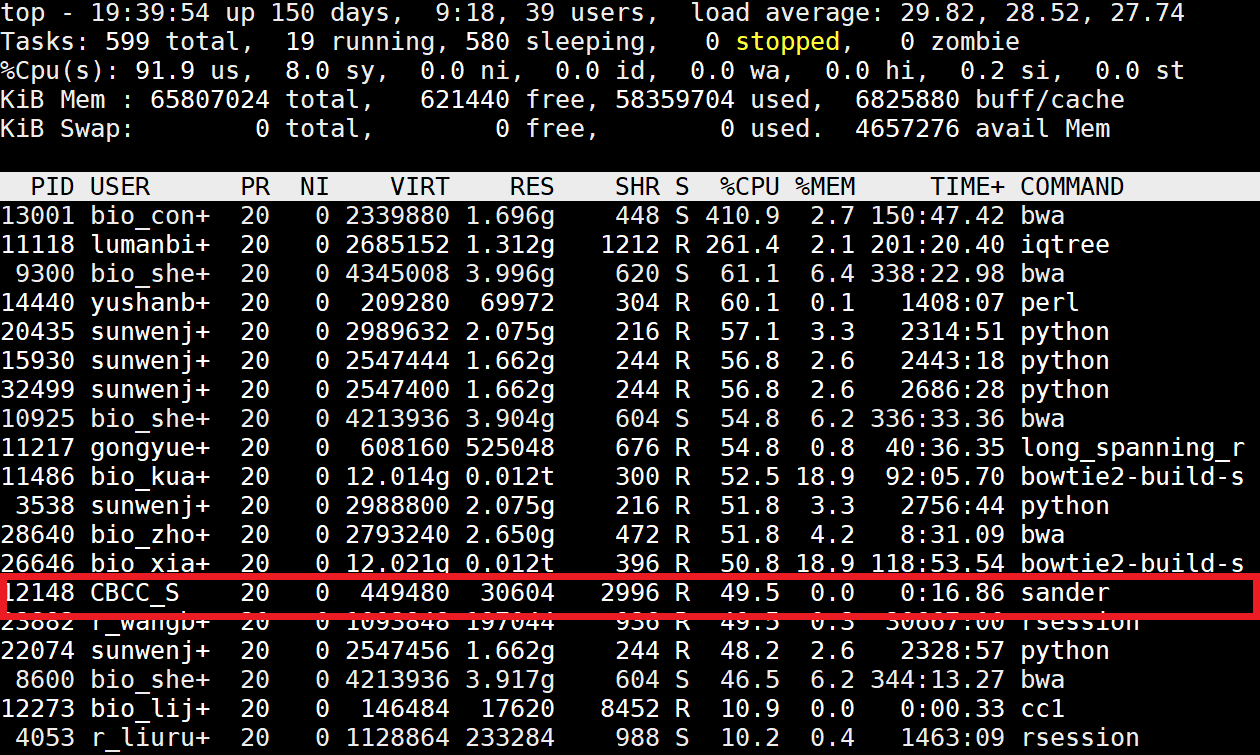 |
Use the top command to view a list of processor tasks and confirm that your simulation is running. You will see a SANDER job with your name using ~100% of the computer's processing power if the simulation is up. Type q to quit the view of processor tasks. $ top Log out of your workstation, log back in, and view the list of processor tasks again to make sure that the nohup command worked and that your simulation is still running. $ top |